HTSeq-Hadoop‘s documentation¶
HTSeqCount¶
The HTSeqCount pipeline is a Hadoop Map-Reduce implementation for the htseq-count Python utility, apart of the HTSeq package, aimed to count the number of short reads from SAM file, mapped to each feature in the GFF or GTF file. For the funcionality description of the htseq-count please refer to the documentation.
Use on Hadoop cluster:
- the mapper HTSeqCount_mapper.py and the reducer HTSeqCount_reducer.py are being supplied to the Hadoop via the -file option
- the JAR-archived General Feature File (GFF) is located on the HDFS and is being distributed to all Hadoop cluster nodes using the Hadoop distribured cache, the -archives option
- Counts from all the Hadoop nodes are summed up during the Reduce stage to produce the final counts
- a use example is in the Running the HTSeqCount on Hadoop section
Use on Linux multicore workstation (cluster node) with GNU parallel:
- input file in SAM format is being filtered if needed and piped from the local filesystem to the STDIN of the HTSeqCount_mapper.py, the local folder with GFF file is specified following the manual
- the mapper produces feature counts for each thread, and piped to the STDIN of the HTSeqCount_reducer.py, which gathers the local counts to the global one.
- a use example is in the Running the HTSeqCount locally section
At the present moment the HTSeqCount is GFF format oriented. More information abput GFF vs. GTF is here: htseq-count FAQ.
HTSeqCount_mapper¶
Should be used in pair with the HTSeq_Hadoop.HTSeqCount_reducer.
Input parameters¶
- -m, --mode (union,intersection-strict,intersection-nonempty)¶
mode to handle reads overlapping more than one feature. Default is union
- -t, --type¶
feature type (3rd column in GFF file) to be used, all features of other type are ignored (default, suitable for GFF files: exon) default=’exon’
- -i, --iaddr¶
GFF attribute to be used as feature ID (default, suitable for Ensembl GTF files: gene_id),default=’gene_id’
- -q, --quite¶
Suppress progress report and warnings
- -gff¶
Folder with the GFF file.
Note
Folder, not a file due to Hadoop distributed cache functionality. Applied for both – local and Hadoop modes.
HTSeqCount_reducer¶
Should be used in pair with the HTSeq_Hadoop.HTSeqCount_mapper.
Running the HTSeqCount on Hadoop¶
The input *.sam.bz2, *.GFF.JAR, and output folder are on the HDFS.
The mapper and reducer are on one local machine, which is a part of the Hadoop cluster.
$HADOOP_HOME/bin/hadoop jar /path/To/hadoop-streaming.jar \
-Dmapred.reduce.tasks=1
-Dmapred.reduce.slowstart.completed.maps=1.0
-mapper /local/path/HTSeqCount_mapper.py -gff ./GFF --type=gene --idattr=ID
-reducer /local/path/HTSeqCount_reducer.py
-archives /hdfs/path/toGFF.JARfolder#GFF
-input /hdfs/folder/input
-output /hdfs/folder/output
-file /local/path/HTSeqCount_mapper.py
-file /local/path/HTSeqCount_reducer.py
Running the HTSeqCount locally¶
A SAM.bz2 file is being uncompressed in parallel with pbzip2, and fed to the STDIN of the GNU Parallel, entries with “mitochondria” and “choloplast” are being filtered out. The remained entries are being split in chunks of 100M and fed to multiple copies of the HTSeq_Hadoop.HTSeqCount_mapper script, which counts the number of times the corresponding genes were ovarlapped by the alingments in each chunk, according to the given policy. The counts from all the copies are sorted and merged to be fed to the HTSeq_Hadoop.HTSeqCount_reducer script to produce the global count file. The intermediate files are being deleted.
f=file.sam.bz2; pbzip2 -dc $f |parallel -k --pipe grep -E -v -i 'mitochondria\|chloroplast' | \
parallel --no-notice --pipe --files --block 100M ./HTSeqCount_mapper.py -gff FOLDER_with_GFF_file -t gene -i ID | \
parallel -Xj1 --no-notice sort -m {} ';' rm {}|
./HTSeqCount_reducer.py >./$f.counts
HTSeqQA¶
The HTSeqQA pipeline is a Hadoop Map-Reduce implementation for the htseq-qa Python utility, the part of the HTSeq package. The purpose of this pipeline is to significantly reduce the quality assessment timing for large datasets in SAM or FASTQ formats, due to massively parallel execution of the code on Hadoop cluster, or on the multicore workstation as an alternative.
For the funcionality description of the htseq-qa please refer to the documentation.
Use on Hadoop cluster
- the mapper HTSeqQA_mapper.py and the reducer HTSeqQA_reducer.py are being supplied to the Hadoop via the -file option
- the dataset of interest in sam.bz2 format is located on the HDFS
- each copy of the mapper operates on one chunk of data and produces its contibution to the global result
- a single copy of the reducer collects the output from each mapper in order to produce a quality assessment picture for the whole dataset in SVG format
- a use example is in the Running the HTSeqQA on Hadoop section
Use on Linux multicore workstation (cluster node) with GNU parallel:
- input file in SAM format is being split into chunks by GNU parallel
- each chunk is piped to the STDIN of a separate copy of the HTSeqQA_mapper.py, which produces and emits its contribution to the global result to the STDOUT
- the output of each mapper is being sorted in a proper way and fed to the STDIN of the single copy of the HTSeqQA_reducer.py, which merges the local contributions to the global one and produces a SVG plot
- a use example is in the Running the HTSeqQA locally section
HTSeqQA_mapper¶
Input parameters¶
- -t, --type¶
Type of file to work on: SAM (default) or FASTQ.
For the real use, the splittable archived input file is on the HDFS. Formats like bzip2 or splittable LZO are either natively supported by Hadoop or this functionality can be easily edded.
- -n, --nosplit¶
Do not split reads in unaligned and aligned ones. Have not tested yet...
Output¶
The mapper emerges the <key,value> pairs, where key is a string, corresponding to the line is the corresponding matrix, and the value is the actual line of matrix.
Details about the matrixes can be found in the HTSeq documentation.
base_arr_U_0 211 158 120 253 0
qual_arr_U_0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 9 0 0 0 0 0 0 8 0 6 12 5 8 0 50 227 0 73 343 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
base_arr_U_1 249 125 121 247 0
qual_arr_U_1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 5 0 0 0 0 0 5 0 0 0 0 0 1 5 0 2 23 13 11 0 37 187 0 30 423 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
base_arr_U_2 227 140 138 237 0
HTSeqQA_reducer¶
Note
The Matlotlib 1.1.1rc does not work with the PDF in the Hadoop framework, while working OK on the local Linux box. Therefore the SVG format was picked up.
Output¶
For the SAM input file, the reducer produces a picture in the SVG format with the contents similar to:
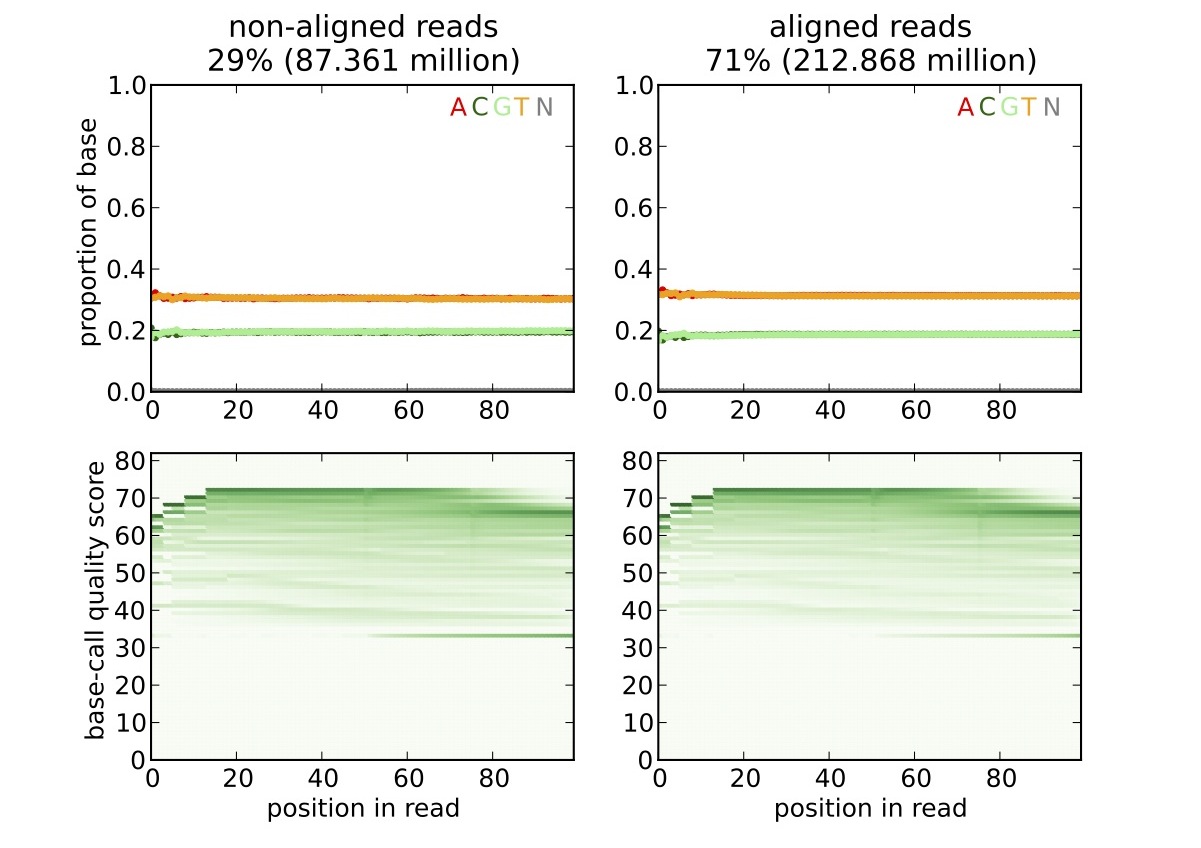
Running the HTSeqQA on Hadoop¶
$HADOOP_HOME/bin/hadoop jar /path/To/hadoop-streaming.jar \
-Dmapred.reduce.tasks=1
-Dmapred.reduce.slowstart.completed.maps=1.0
-mapper /local/path/HTSeqQA_mapper.py -m 80
-reducer /local/path/HTSeqQA_reducer.py
-input /hdfs/folder/input
-output /hdfs/folder/output
-file /local/path/HTSeqQA_mapper.py
-file /local/path/HTSeqQA_reducer.py
Running the HTSeqQA locally¶
The Hadoop pipeline can be tested locally on a Linux box as a single-threaded application:
bzcat file.sam.bz2 | ./HTSeqQA_mapper.py -m 70 | sort - k1,1 | ./HTSeqQA_reducer.py >tmp.svg
which will produce the tmp.svg file with the results.
Another option is to use GNU Parallel and run the Mapper and Reducer on the multicore node:
pbzip2 -dc $file.sam.bz2 | \
parallel --no-notice --pipe --block 100M \
./HTSeqQA_mapper.py -m 80 2>/dev/null | \
parallel --pipe --files sort -t"_" -k1,3 -k4,4n | \
parallel -Xj1 --no-notice sort -m -t"_" -k1,3 -k4,4n {} ';' rm {} | \
./HTSeqQA_reducer.py >pic.svg
Brief description: pbzip2 is a parallel version of bzip2 archiver. The pipeline unarchives the file.sam.bz2, cuts it into 100M chunks and feeds these chunks to the HTSeqQA_mapper.py. The output of each mapper is being sorted according to the syntaxis of the mapper, and merged afterwards, the temporary files are deleted. The prepared data comes to the STDIN of the HTSeqQA_reducer.py to produce pic.svg.
Indices and tables¶
Contents¶
Author¶
Please contact Alexey Siretskiy if needed.